








Thứ 5 ngày 04 tháng 10 năm 2018Lượt xem: 16238
Liệt trên nhân tiến triển.
Liệt trên nhân tiến triển (PSP – Progressive supranuclear palsy) là bệnh lý thoái hóa thần kinh phổ biến nhất trong nhóm hội chứng Parkinson không điển hình. Các triệu chứng lâm sàng đặc trưng của bệnh gồm liệt liếc dọc, mất ổn định tư thế với tình trạng ngã tái diễn không rõ nguyên nhân, giảm động và rối loạn nhận thức. Về mặt bệnh lý, liệt trên nhân tiến triển được đặc trưng bởi sự lắng đọng protein TAU bất thường và được xếp vào nhóm bệnh lý TAU protein.
1. Lịch sử
Năm 1964, ba tác giả Steele, Richardson và Olszewski là những người đầu tiên đưa ra mô tả về liệt trên nhân tiến triển thông qua 9 ca bệnh với các biểu hiện: liệt vận nhãn trên nhân tiến triển, rối loạn tư thế và dáng đi, rối loạn vận ngôn, nuốt khó, tăng trương lực cơ và suy giảm nhận thức thùy trán. Do vậy, liệt trên nhân tiến triển có tên gọi là hội chứng Steele-Richardson-Olszewski. Kể từ đó, hàng trăm các ca bệnh tương tự đã được ghi nhận. Kiểu hình lâm sàng rối loạn vận động cổ điển hiện được gọi là hội chứng Richardson (PSP-RS).
Năm 1972, Steele dự đoán các biến thể lâm sàng của hội chứng có khả năng xảy ra khi bệnh ảnh hưởng đến các nhân thân não khác nhau tại các thời điểm khác nhau và ở các mức độ khác nhau. Các kiểu hình khác nhau đã được ghi nhận. Một số biến thể của liệt trên nhân tiến triển bao gồm PSP với biểu hiện đông cứng dáng đi tiến triển (PSP-PGF), PSP với hội chứng Parkinson chiếm ưu thế (PSP-P) và PSP với rối loạn chức năng thùy trán chiếm ưu thế (PSP-F).
Tiêu chuẩn liệt trên nhân tiến triển ban đầu được đề xuất vào năm 1996 và được sửa đổi vào năm 2017.
2. Dịch tễ
Liệt trên nhân tiến triển được coi là một căn bệnh hiếm gặp với tỷ lệ hiện mắc khoảng 5,8 đến 6,5 trên 100.000. Tỷ lệ mắc bệnh liệt trên nhân tiến triển hàng năm dao động từ 0,3 đến 0,4 trên 100.000. Tỷ lệ mắc hàng năm tăng theo độ tuổi từ 1,7 trường hợp trên 100.000 ở độ tuổi 50 đến 59 tuổi lên 14,7 trên 100.000 ở 80 đến 89 tuổi.
Tuổi khởi phát trung bình của bệnh liệt trên nhân tiến triển là khoảng 65 tuổi, già hơn so với bệnh Parkinson vô căn. Hầu như không có trường hợp liệt trên nhân tiến triển nào được xác nhận qua khám nghiệm tử thi đã được báo cáo ở những bệnh nhân dưới 40 tuổi. Báo cáo ban đầu ghi nhận tỷ lệ nam giới chiếm ưu thế (8/1). Tuy nhiên, các báo cáo sau đó không tìm thấy ưu thế giới rõ ràng trong bệnh liệt trên nhân tiến triển.
Không có yếu tố nguy cơ nào được chứng minh là có liên quan đến liệt trên nhân tiến triển trừ tuổi cao. Hiện nay có nhiều báo cáo cho thấy tính nhạy cảm về mặt di truyền. Sa sút trí tuệ trán thái dương có hội chứng Parkinson do đột biến gen MAPT (FTDP-17T) có nhiều biến thể lâm sàng và bệnh học, trong đó FTDP-17T do mã hóa hoặc ghép nối exon 10 có nhiều điểm giống liệt trên nhân tiến triển cả về lâm sàng và mô bệnh học. Ngoài ra một số gen khác làm tăng nguy cơ liệt trên nhân tiến triển như STX6, EIF2AK3, MOBP; tuy nhiên ý nghĩa của những gen này còn chưa rõ ràng.
3. Sinh lý bệnh và giải phẫu bệnh
Liệt trên nhân tiến triển thường thấy teo thùy trán (đặc biệt là hồi trước trung tâm), não giữa (đặc biệt là phần mái), cầu não teo ở mức độ nhẹ hơn. Nhân dưới đồi nhỏ hơn và có thể bị chuyển sang màu xám. Phần trên của cuống tiểu não và nhân răng tiểu não thường bị teo và có màu xám do mất sợi myelin.
Đặc điểm mô bệnh học của liệt trên nhân tiến triển bao gồm mất tế bào thần kinh, tăng sinh xơ hóa và mô đệm, thể vùi TAU protein trong tế bào hình sao, tế bào ít nhánh và neuron chủ yếu ở nhân dưới đồi, cầu nhạt, thể vân, nhân đỏ, chất đen, chỏm cầu não, nhân vận nhãn và nhân răng. Các đám rối tơ sợi thần kinh chủ yếu chứa các đồng dạng TAU với bốn lần lặp lại liên kết vi ống (4R-TAU) trong tế bào thần kinh đệm oligodendrocyte và tế bào hình sao. Bình thường, TAU được phosphoryl hóa trên một loạt các gốc serine và threonine, được điều chỉnh bởi nhiều kinase và phosphatase. Trong liệt trên nhân tiến triển, protein TAU bị phosphoryl hóa quá mức dẫn đến mất ái lực với các vi ống làm mất ổn định, tháo rời vi ống. Điều này dẫn đến sự tích tụ của TAU và hình thành các đám rối sợi thần kinh.
Ở bệnh Alzheimer, sự hình thành đám tơ sợi thần kinh do lắng đọng protein TAU xảy ra ở tế bào thần kinh và xuất hiện đầu tiên ở hệ limbic. Trong khi đó ở PSP, sự lắng đọng này xuất hiện cả ở tế bào thần kinh và tế bào thần kinh đệm (bao gồm cả oligodendrocyte và astrocyte), tổn thương xuất hiện đầu tiên ở vỏ não vận động và tiền vận động.
4. Lâm sàng
Đặc điểm ban đầu thường gặp nhất với kiểu hình “cổ điển” phổ biến nhất của liệt trên nhân tiến triển hay hội chứng Richardson (PSP-RS) là rối loạn dáng đi dẫn đến ngã. Liệt vận nhãn trên nhân là dấu hiệu nhận biết của bệnh liệt trên nhân tiến triển. Ngoài ra, liệt trên nhân tiến triển còn có các dấu hiệu lâm sàng khác như nói khó, khó nuốt, liệt giả hành tủy, cứng, giảm động, rối loạn nhận thức thùy trán và rối loạn giấc ngủ. Biểu hiện lâm sàng khá đa dạng với nhiều kiểu hình biến thể khác nhau.
4.1. Mất ổn định tư thế và ngã
Bệnh nhân PSP-RS có dáng đi cứng, chân đế rộng bước chân có độ dài thay đổi, đầu gối và cơ trục thân có xu hướng ở tư thế duỗi (ngược lại với bệnh nhân Parkinson là tư thế gấp), cánh tay hơi dạng ra. Các bất thường này làm bệnh nhân có xu hướng lảo đảo, loạng choạng và còn được gọi là “dáng đi của thủy thủ say rượu” (drunken sailor gait). Thay vì xoay người chậm chạp đưa cả thân mình xoay cùng trục như ở bệnh nhân Parkinson, bệnh nhân PSP có xu hướng xoay người không đồng bộ làm tăng nguy cơ ngã. Bệnh nhân thường ngã ngửa gây ra các chấn thương bầm tím, gãy xương, chảy máu não, thậm chí tử vong.
4.2. Rối loạn vận nhãn
Liệt vận nhãn trên nhân là đặc điểm lâm sàng đặc trưng của liệt trên nhân tiến triển. Biểu hiện đầu tiên là mất cử động mắt nhanh (đặc điểm quan trọng giúp chẩn đoán sớm), sau đó là hạn chế vận nhãn theo chiều dọc và chức năng nhìn xuống thường bị ảnh hưởng sớm hơn chức năng nhìn ngang. Có thể kèm theo hạn chế nhìn liếc ngang. Các triệu chứng bất thường về cử động mắt xuất hiện trung bình 4 năm sau khi bệnh khởi phát. Ban đầu triệu chứng liệt vận nhãn có thể được bù trừ bằng phản xạ mắt búp bê (Doll’s eyes) nhưng khi bệnh tiến triển liên quan đến thân não thì phản xạ tiền đình mắt sẽ mất đi. Ở giai đoạn cuối của bệnh, mắt hầu như bất động.
Các dấu hiệu vận nhãn khác bao gồm: cử động nhãn cầu dạng giật sóng vuông (square wave jerks) khi nhìn cố định, co thắt mi mắt, mất hội tụ nhãn cầu. Giảm chớp mắt, loạn trương lực cơ mặt, bất thường vận nhãn làm khuôn mặt bệnh nhân luôn biểu hiện như “sự ngạc nhiên”. Hạn chế liếc dọc thường làm cho bệnh nhân khó khăn khi đọc, ăn uống và đi lại dễ té ngã.
4.3. Các triệu chứng vận động
Hội chứng Parkinson ở tất cả các thể của liệt trên nhân tiến triển đều đặc trưng bởi giảm động và chữ viết nhỏ. Liệt trên nhân tiến triển thường có biểu hiện cứng cơ ở trục thân hơn ở chi, đặc biệt là cơ cổ và phần trên của thân mình. Loạn trương lực cơ cổ có thể xuất hiện đặc biệt là vẹo cổ ra sau (retrocollis). Ngoài ra còn có thể gặp co thắt mi mắt, loạn trương lực cơ nửa người thường hiếm gặp.
Khoảng 1/3 số bệnh nhân liệt trên nhân tiến triển có dấu hiệu tháp bao gồm tăng phản xạ gân xương và dấu hiệu Babinski. Dấu hiệu gan tay cằm và các dấu hiệu giải phóng chức năng thùy trán có thể gặp. Lưỡi bệnh nhân bị co rút và vận động chậm. Rối loạn vận ngôn, khó phát âm, khó nuốt gặp ở giai đoạn giữa và sau của bệnh. Ngoài ra có thể có nói lắp và lặp từ.
Một số ít bệnh nhân có thể đáp ứng với liệu pháp dopaminergic trong giai đoạn đầu cảu bệnh nhưng phần đa là không đáp ứng.
4.4. Suy giảm nhận thức
Suy giảm nhận thức của liệt trên nhân tiến triển chủ yếu liên quan đến rối loạn chức năng thùy trán. Các bệnh nhân suy giảm suy nghĩ trừu tượng, giảm khả năng nói trôi chảy, các cử động lặp lại dù không còn kích thích và rối loạn hành vi thùy trán. Ở giai đoạn sau của bệnh, rối loạn chức năng điều hành biểu hiện rõ.
4.5. Các rối loạn khác
Các bất thường khác có thể gặp như thờ ơ, giải ức chế, trầm cảm, lo âu, rối loạn ám ảnh cưỡng chế.
Liệt giả hành tủy là một biểu hiện đặc trưng khác của liệt trên nhân tiến triển.Bệnh nhân thường có giọng nói khàn đặc trưng và không kiểm soát được cảm xúc (ít gặp hơn so với liệt giả hành tủy do các nguyên nhân khác).
Liệt trên nhân tiến triển có thể gặp mất ngủ, khó duy trì giấc ngủ. rối loạn hành vi trong giấc ngủ giai đoạn động mắt nhanh (RBD) ít khi gặp.
5. Cận lâm sàng
5.1. Hình ảnh học
* Chụp cộng hưởng sọ não.
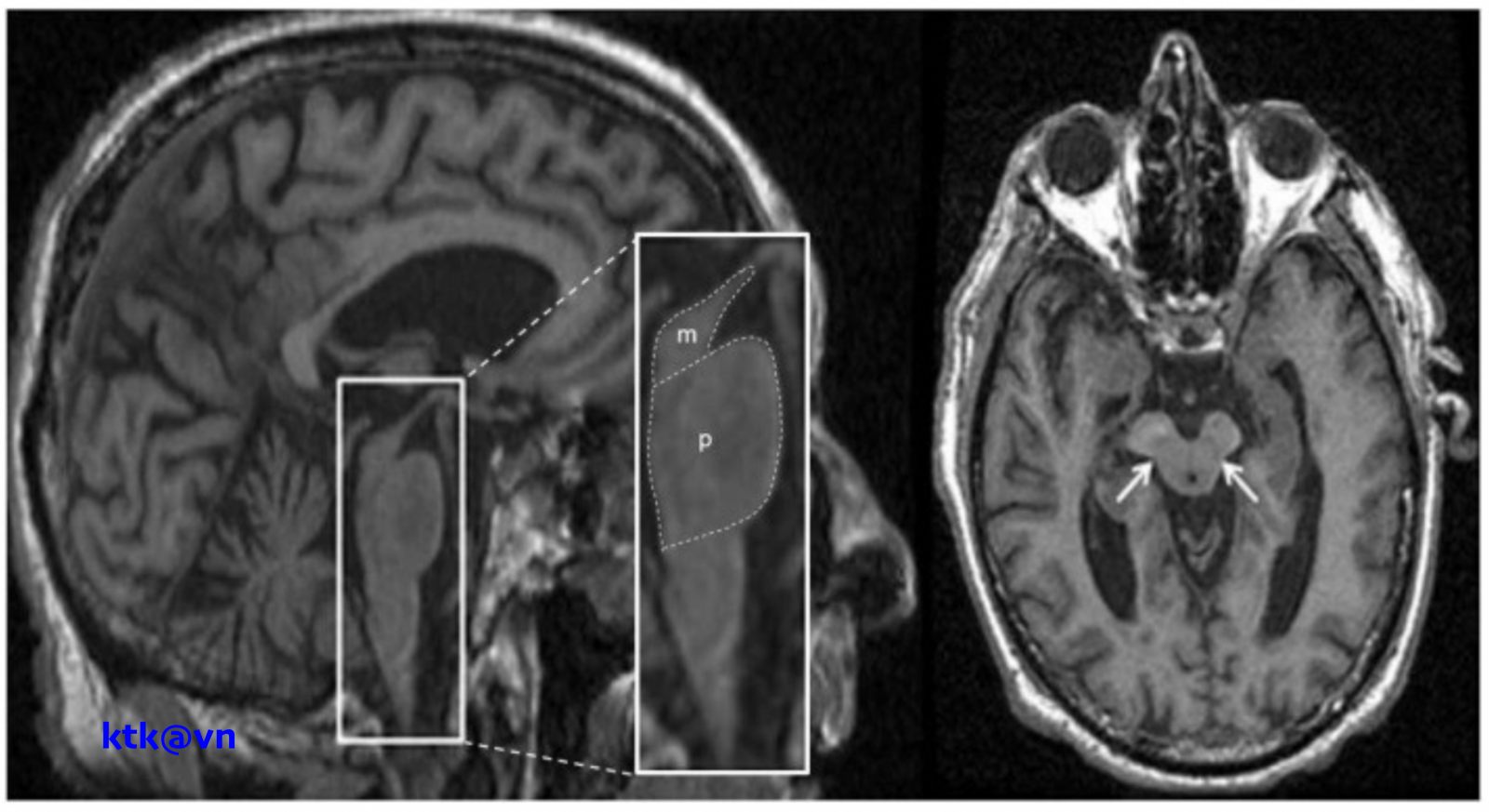
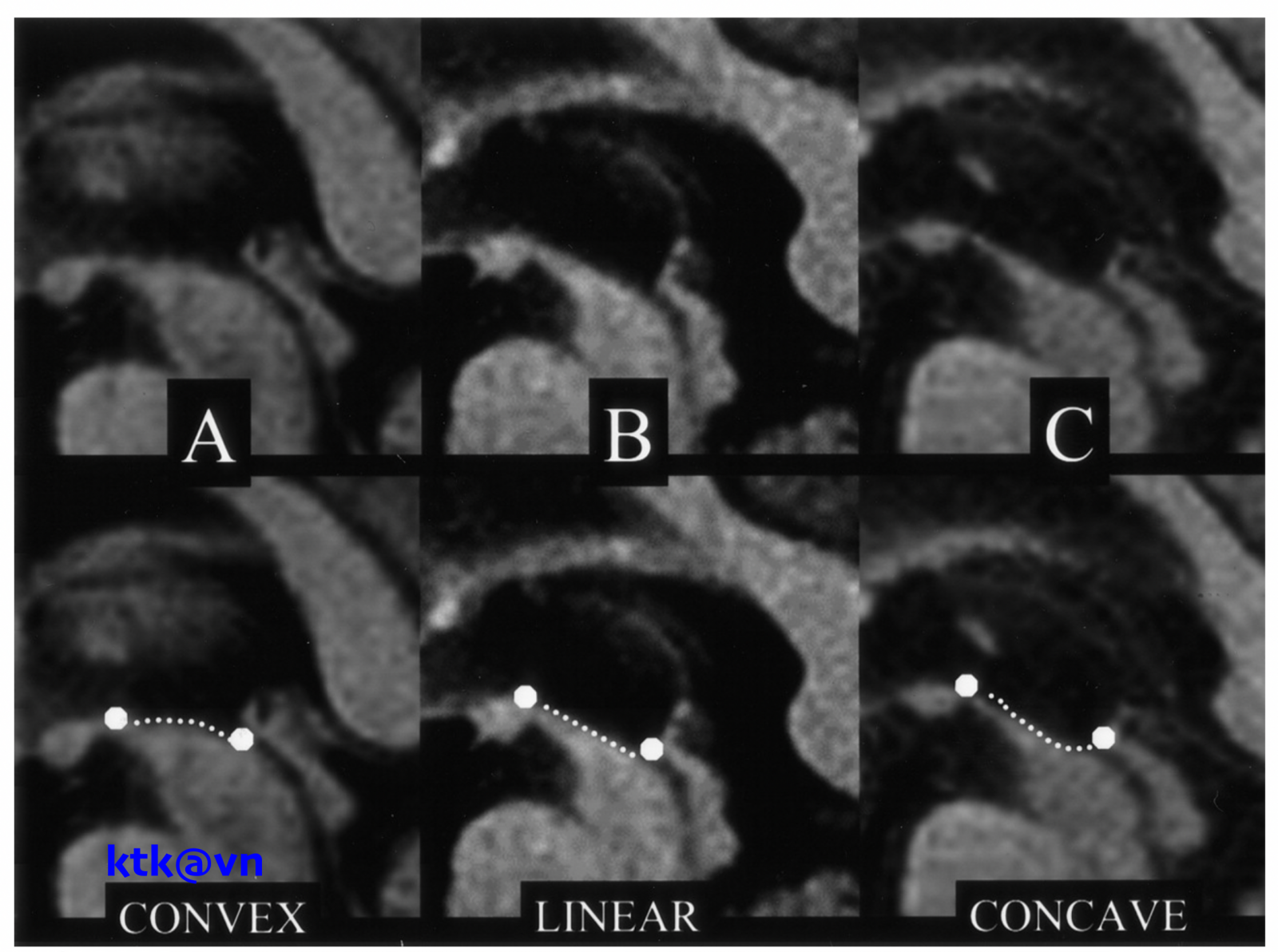
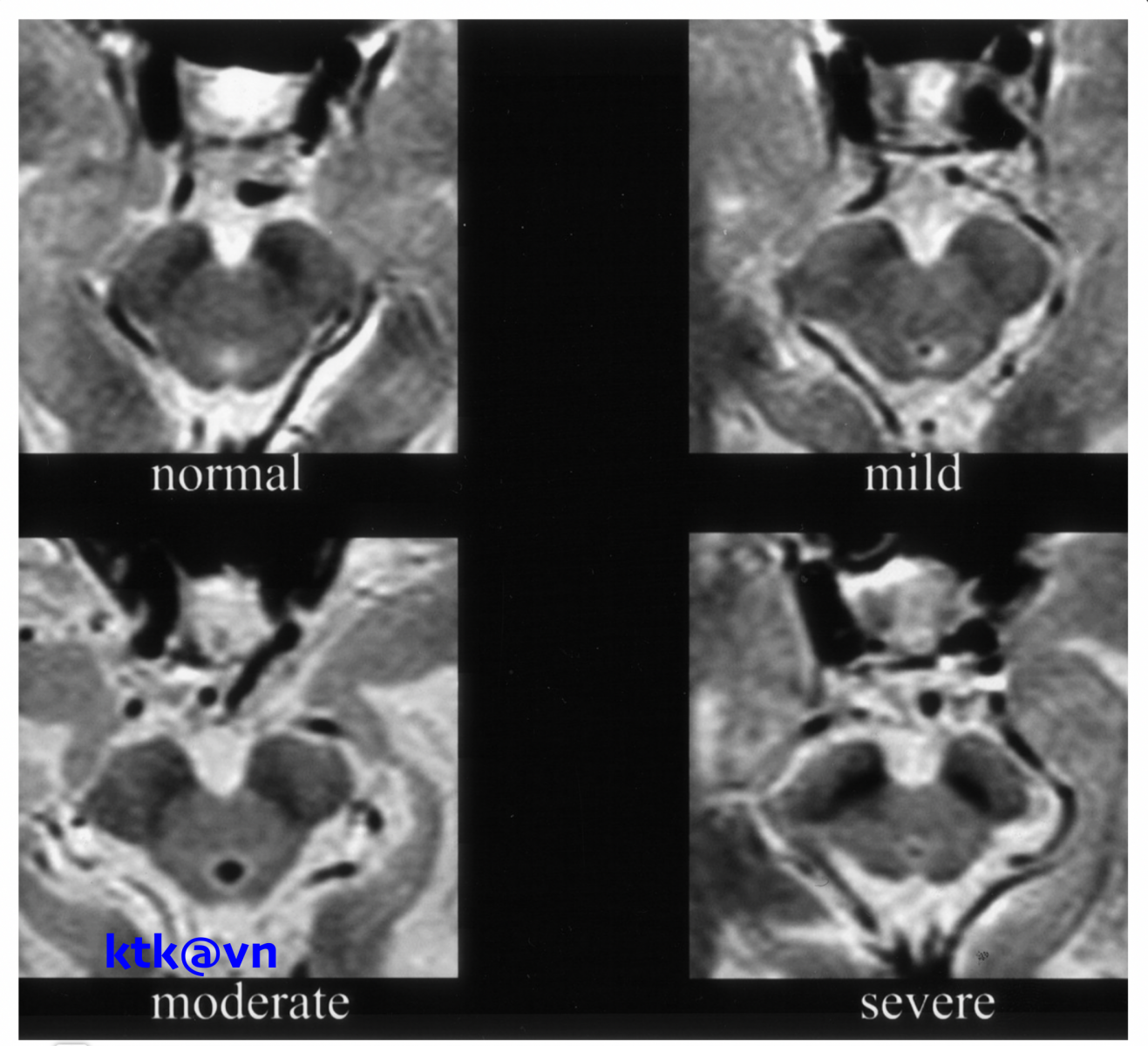
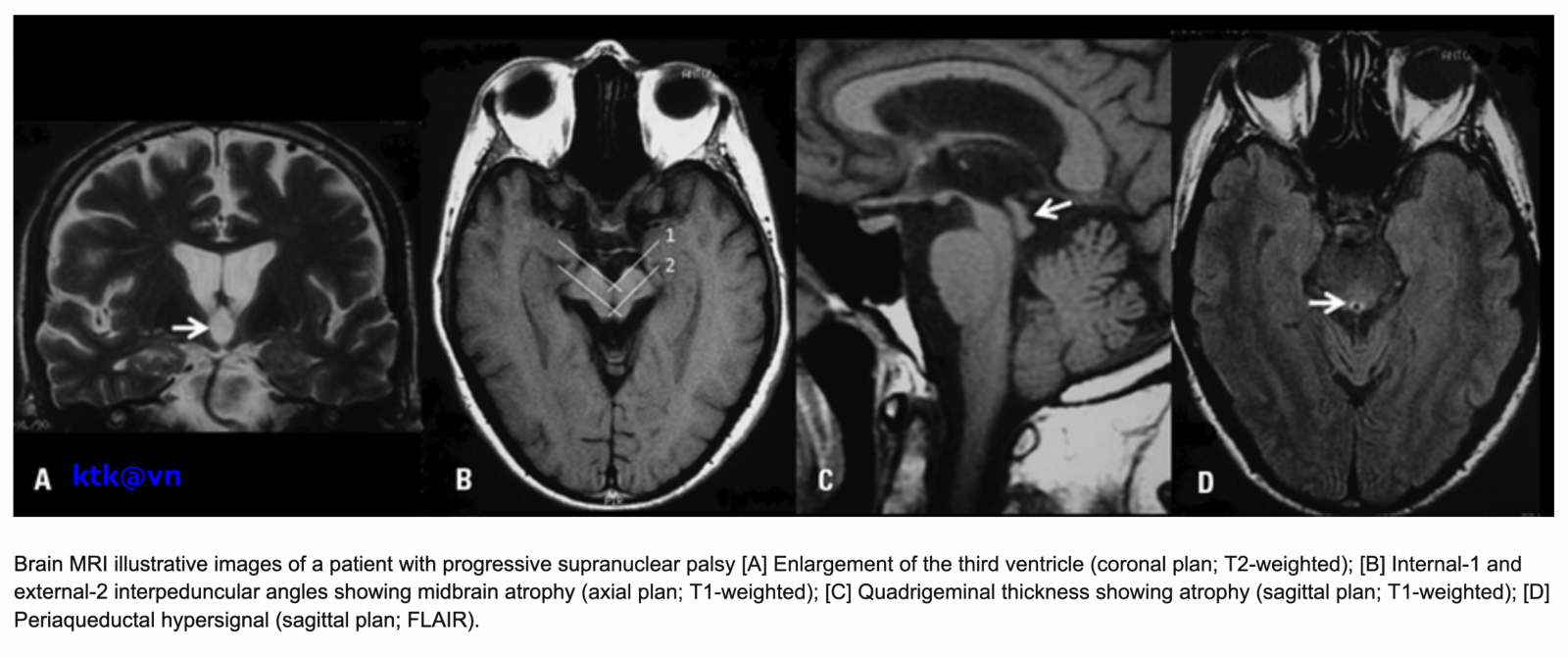
Những đặc điểm thường gặp trên hình ảnh phim chụp cộng hưởng từ sọ não là teo não giữa, giãn não thất ba, teo cuống tiểu não trên (SCP), teo thùy trán và thái dương. Teo não giữa được thể hiện trực quan qua hình ảnh “mỏ chim ruồi” khi nhìn trên lát cắt đứng dọc, hình ảnh “chuột Mickey” khi nhìn trên lát cắt ngang, hình ảnh “morning glory” (một số tài liệu gọi là dấu hiệu hoa mòng biển) trên lát cắt ngang. Teo cuống tiểu não trên, dấu hiệu “chim ruồi”, dấu hiệu “hoa mòng biển” trên cộng hưởng từ cấu trúc có độ đặc hiệu cao nhưng độ nhạy kém hơn tiêu chuẩn lâm sàng.
Các phép đo định lượng não giữa, cuống tiểu não và các cấu trúc thân não khác, đã cho thấy độ nhạy và độ đặc hiệu cao hơn:
– Đo lường chiều rộng trước sau và thể tích của cuống não của bệnh nhân PRP-RS nhỏ hơn bệnh nhân teo đa hệ thống và bệnh Parkinson. Liệt trên nhân tiến triển có giảm diện tích não giữa trên mặt phẳng đứng dọc giữa, giảm tỷ lệ cuống não/cầu não. Tuy nhiên độ nhạy và độ đặc hiệu của tỷ lệ cuống não/cầu não phân biệt PSP-RS và teo đa hệ thống thể Parkinson (MSA-P) còn dao động khác nhau giữa các nghiên cứu.
– Chỉ số parkinson cộng hưởng từ (MRPI) [= diện tích cầu não /não giữa) * (chiều rộng cuống tiểu não giữa/cuống tiểu não trên)] có độ nhạy và độ đặc hiệu cao trong chẩn đoán phân biệt liệt trên nhân tiến triển với MSA-P và bệnh Parkinson, tuy nhiên để tính được chỉ số này yêu cầu một phương pháp đo chi tiết, khó phổ biến cho các trung tâm và đưa vào thường quy.
* Chụp PET/ SPECT
Chụp FDG-PET (18F fluorodeoxy glucose), liệt trên nhân tiến triển có hình ảnh giảm chuyển hóa ở não giữa là sớm nhất sau đó ở hạch nền, đồi thị, thùy trán, chủ yếu vùng trước vận động, có độ nhạy 93% và độ đặc hiệu 90% khi phân biệt PSP-RS với bệnh Parkinson, teo đa hệ thống và thoái hóa vỏ hạch nền. TAU-PET với chất đánh dấu liên kết chọn lọc với TAU-protein là một công cụ đầy hứa hẹn cho phép quan sát phân bố đám rối sợi thần kinh. TAU-PET giúp đánh giá tương quan giữa hình ảnh và giai đoạn bệnh. Các nghiên cứu TAU-PET có nhiều hứa hẹn tuy nhiên mới được thực hiện trong nghiên cứu ở những nhóm nhỏ và chưa áp dụng lâm sàng.
Chụp SPECT (chụp cắt lớp vi tính bằng bức xạ đơn photon) sử dụng chất đánh dấu dopamin hoạt động cho thấy sự giảm hấp thu chất đánh dấu trong thể vân.
Một số hình ảnh nhận biết Liệt trên nhân tiến triển (Phim chụp cộng hưởng từ (MRI) sọ não)
5.2. Các dấu ấn sinh học
Các dấu ấn sinh học trong dịch não tủy trong bệnh liệt trên nhân tiến triển vẫn đang được nghiên cứu phát triển. TAU protein toàn phần (t-TAU) và TAU protein phosphoryl hóa (p-TAU) giảm trong dịch não tủy của bệnh nhân liệt trên nhân tiến triển so với nhóm chứng, điều này ngược lại ở bệnh nhân Alzheimer. Tuy nhiên có sự chồng lấp đáng kể và những dấu ấn sinh học này không được sử dụng để phân biệt liệt trên nhân tiến triển với nhóm chứng cũng như các bệnh khác của TAU protein.
Protein chuỗi nhẹ Neurofilament (NfL) – một dấu ấn do sự phá hủy sợi trục có thể là dấu ấn hữu ích ở liệt trên nhân tiến triển và các bệnh lý thần kinh khác, nhưng không phải là dấu ấn có tính chất chẩn đoán bệnh liệt trên nhân tiến triển. NfL trong dịch não tủy ở bệnh nhân liệt trên nhân tiến triển và hội chứng Parkinson không điển hình cao hơn so với bệnh Parkinson và nhóm chứng. NfL cũng tăng trong máu ngoại vi và mức độ tăng tương ứng với mức độ trong dịch não tủy.
6. Tiêu chuẩn chẩn đoán
Năm 2017, ội rối loạn vận động (MDS) đã đưa ra tiêu chuẩn chẩn đoán mới cho bệnh liệt trên nhân tiến triển. Tiêu chuẩn chẩn đoán này bao gồm các phần:
– Các đặc điểm cơ bản: bao gồm tiêu chuẩn bắt buộc và tiêu chuẩn loại trừ cần thiết cho chẩn đoán.
– 4 lĩnh vực chức năng cốt lõi (rối loạn vận nhãn, mất ổn định tư thế, giảm động và suy giảm nhận thức): là các biểu hiện đặc trưng của liệt trên nhân tiến triển.
– Các tiêu chuẩn hỗ trợ tăng độ tin cậy cho chẩn đoán.
– Định nghĩa chi tiết các tiêu chuẩn cốt lõi và tiêu chuẩn hỗ trợ.
– 4 mức độ chắc chắn chẩn đoán.
Mời xem Tiêu chuẩn Chẩn đoán Liệt trên nhân tiến triển.
7. Chẩn đoán thể bệnh
Tiêu chí MDS-PSP xác định các loại ưu thế lâm sàng dựa trên các đặc điểm lâm sàng, bao gồm:
– PSP với hội chứng Richardso. (PSP-RS)
– PSP với hội chứng parkinson chiếm ưu thế (PSP-P)
– PSP với rối loạn chức năng vận nhãn chiếm ưu thế (PSP-OM)
– PSP với mất ổn định tư thế chiếm ưu thế (PSP-PI)
– PSP với đông cứng dáng đi tiến triển (PSP-PGF)
– PSP với rối loạn chức năng thùy trán chiếm ưu thế (PSP-F)
– PSP với chứng rối loạn lời nói/ngôn ngữ chiếm ưu thế (PSP-SL)
– PSP với hội chứng vỏ não hạch nền chiếm ưu thế (PSP-CBS).
Tiêu chuẩn chẩn đoán MDS-PSP đã bỏ qua hai kiểu hình biến thể hiếm được công nhận khác (PSP với chứng mất điều hòa tiểu não chiếm ưu thế [PSP-C] và PSP với bệnh xơ cứng teo cơ bên nguyên phát chiếm ưu thế [PSP-PLS]) vì bằng chứng bệnh học lâm sàng không đủ để đưa ra các lâm sàng tiêu chuẩn chẩn đoán đủ độ đặc hiệu.
8. Chẩn đoán phân biệt
– Bệnh Parkinson: khởi phát không đối xứng, ngã và nuốt khó xảy ra ở giai đoạn từ giữa đến tiến triển, đáp ứng với levodopa, tiến triển chậm hơn so với liệt trên nhân tiến triển.
– Thoái hóa vỏ não hạch nền: bệnh nhân thường có mất thực dụng động tác, loạn trương lực cơ, giật cơ, hội chứng bàn tay người ngoài hành tinh, mất cảm giác vỏ não..
– Teo đa hệ thống: rối loạn thần kinh tự chủ và hội chứng tiểu não chiếm ưu thế trong khi nhận thức được bảo tồn.
– Bệnh Pick, bệnh Alzheimer: 2 bệnh này thường là sa sút trí tuệ kiểu vỏ não trong khi liệt trên nhân tiến triển thường gặp sa sút trí dưới vỏ.
9. Điều trị
Hiện tại chưa có phương pháp điều trị nào có thể chữa được bệnh. Mục đích của điều trị là cải thiện triệu chứng và chất lượng cuộc sống cho bệnh nhân.
9.1. Điều trị triệu chứng
Hội chứng Parkinson, rối loạn tư thế và ngã: Levodopa được điều trị thử cho bệnh nhân chậm vận động và tăng trương lực cơ, ngã. Sử dụng Levodopa tăng dần liều đến 1000 mg/ngày và duy trì ít nhất một tháng. Liệt trên nhân tiến triển thường không có đáp ứng hoặc đáp ứng kém với Levodopa ở giai đoạn đầu của bệnh. Thể PSP-P ban đầu thường đáp ứng, trong khi các thể khác của liệt trên nhân tiến triển thì không. Các thuốc đồng vận Dopamin và ức chế MAO-B thường ít hiệu quả và không được dùng cho bệnh nhân liệt trên nhân tiến triển. Amantadin với liều tăng chậm, đến 100-300 mg/ngày có thể cải thiện triệu chứng vận động chậm và tăng trương lực cơ, cũng như đông cứng dáng đi, cải thiện thăng bằng. Có thể dùng đơn độc hoặc kết hợp với Levodopa.
Các rối loạn vận động khác như loạn trương lực cơ cổ, loạn trương lực cơ ở chi, co thắt mi mắt có thể đáp ứng tốt với tiêm độc tố Botulinum.
Clonazepam hoặc Levitiracetam có thể được sử dụng cho giật cơ trong hội chứng vỏ não hạch nền.
DBS hiện không được khuyến cáo cho bệnh liệt trên nhân tiến triển bên ngoài cơ sở nghiên cứu.
Rối loạn nhận thức: không có điều trị đặc hiệu cho rối loạn nhận thức ở bệnh nhân liệt trên nhân tiến triển. Thực tế là thuốc ức chế enzym cholinesterase không được khuyến cáo do có khả năng làm xấu hơn triệu chứng vận động.
Các triệu chứng khác:
– Các thuốc ức chế tái hấp thu Serotonin chọn lọc có hiệu quả trong điều trị trầm cảm, rối loạn lo âu, rối loạn cảm xúc. Ngoài ra, Trazodone và Mirtazapine cũng đã được báo cáo có tác dụng điều trị ở một số bệnh nhân. Zolpidem có thể sử dụng để điều trị mất ngủ.
– Không có thuốc điều trị khó nuốt. Ở giai đoạn sau của bệnh có thể phải cho ăn qua mở thông dạ dày.
– Chảy nước dãi có thể kiểm soát bằng Atropine 1% nhỏ tại chỗ, thận trọng với các tác dụng phụ. Có thể tiêm độc tố Botulinum vào tuyến nước bọt nhưng có thể làm nặng thêm khó nuốt.
Vật lý trị liệu và vận động, liệu pháp ngôn ngữ, tập nuốt: có vai trò quan trọng để ngăn ngừa té ngã, duy trì khả năng vận động, ngôn ngữ và cải thiện các hoạt động sinh hoạt hàng ngày.
9.2 Các thuốc thay đổi bệnh
Cho đến nay, các nghiên cứu về các liệu pháp điều chỉnh bệnh có khả năng không chứng minh được hiệu quả ở những người bị nghi ngờ mắc bệnh liệt trên nhân tiến triển. Các thử nghiệm ngẫu nhiên, có đối chứng với giả dược về riluzole, davunetide, waveglusib, sodium valproate và rasagiline cho thấy không có tác động đến các tiêu chí chính theo dõi sự tiến triển của bệnh và thiếu bằng chứng cho thấy các tác nhân có tác dụng dự kiến thông qua các cơ chế lý thuyết.
Các nghiên cứu hiện tại về liệu pháp điều trị liệt trên nhân tiến triển tập trung vào TAU bao gồm TPI-287 (chất ổn định vi ống), C2N-8E12/ABBV-8E12 và BMS-986168/BIIB092 (các kháng thể đơn dòng chống TAU và salsalate, chất ức chế acetyl hóa TAU). Các chất ổn định vi ống được hy vọng sẽ bù đắp cho rối loạn chức năng vi ống liên quan đến mất chức năng TAU; các kháng thể đơn dòng chống TAU được hy vọng sẽ ngăn cản sự lây lan của TAU gây bệnh, và các chất ức chế acetyl hóa TAU được hy vọng sẽ ức chế sự acetyl hóa của TAU hòa tan và do đó hạn chế sự tăng phosphoryl hóa.
10. Tiến triển và tiên lượng
Liệt trên nhân tiến triển tiến triển nhanh và không ngừng. Hầu hết bệnh nhân trở nên phụ thuộc vào sự chăm sóc trong vòng 3 hoặc 4 năm kể từ khi khởi phát. Tử vong trung bình từ 6 đến 9 năm sau khi được chẩn đoán.
Tin xem nhiều nhất
-
-
Ngày 09/02/2018
ĐIỆN CƠ là gì ...
-

-
Ngày 13/02/2018
Điều trị Co thắt mi mắt (Blepharospasm)?
-

-
Ngày 01/03/2018
Điều trị co cứng cơ sau Đột quỵ não.
-
-
Ngày 05/10/2021
Chẩn đoán định khu tổn thương tủy sống.
-

-
Ngày 05/04/2020
Liệt dây thần kinh số VII.

